
Monogenic and Syndromic
Obesity
Sonali Malhotra, MD
Pediatric Endocrinology/Obesity Medicine
Diplomate, ABOM
Harvard Medical School
Massachusetts General Hospital for Children
Disclosures
Speaker Bureau: Rhythm Pharmaceuticals

Learning Objectives
• Review the energy balance regulation pathway.
• To introduce rare disorders of obesity, also known as monogenic
obesity.
• Review clinical features of syndromic genetic disorders that cause
obesity
Objectives
• To discuss the energy balance regulation
• To introduce rare disorders of obesity, also known as monogenic
obesity.
• Review clinical features of syndromic genetic disorders that cause
obesity
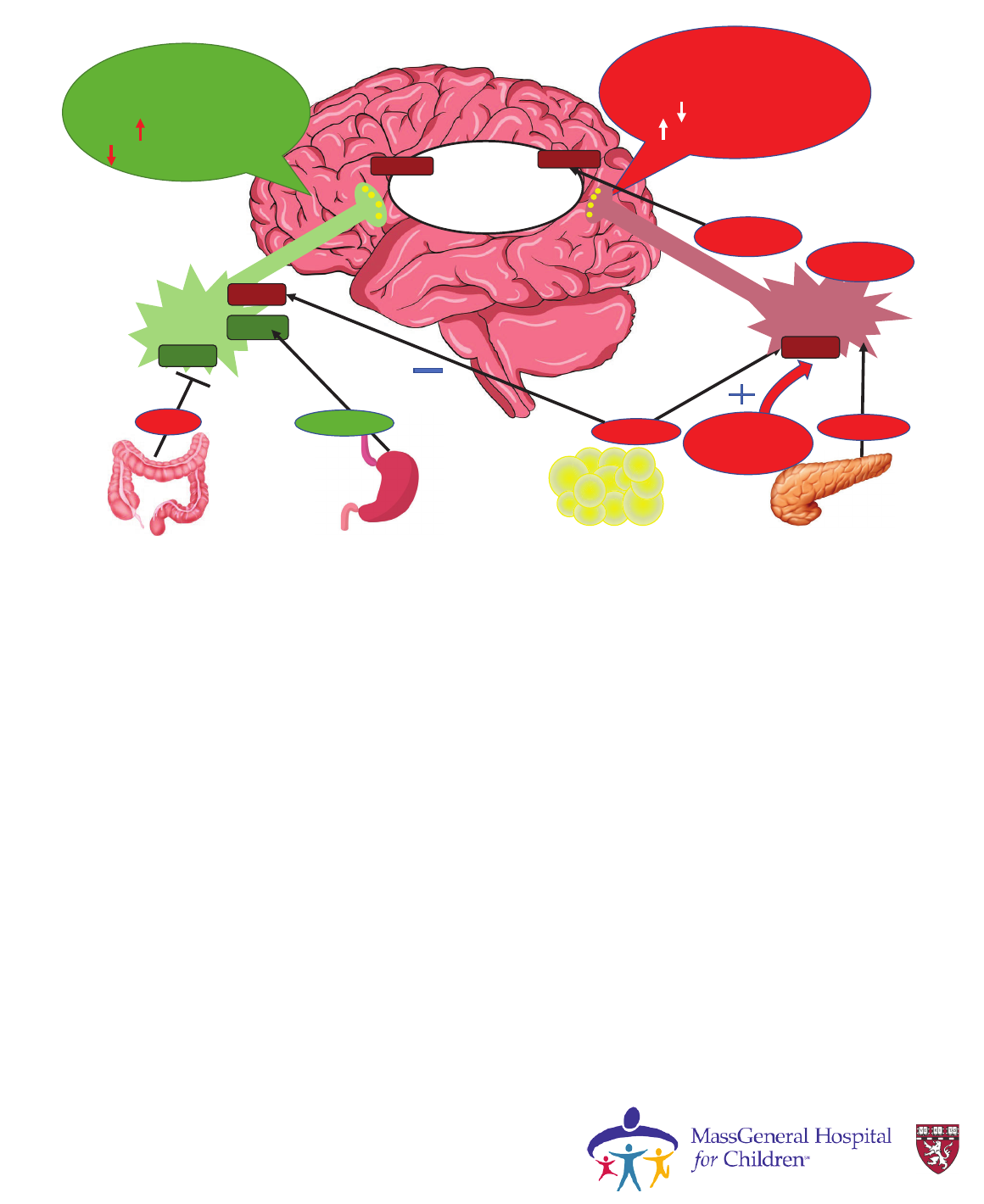
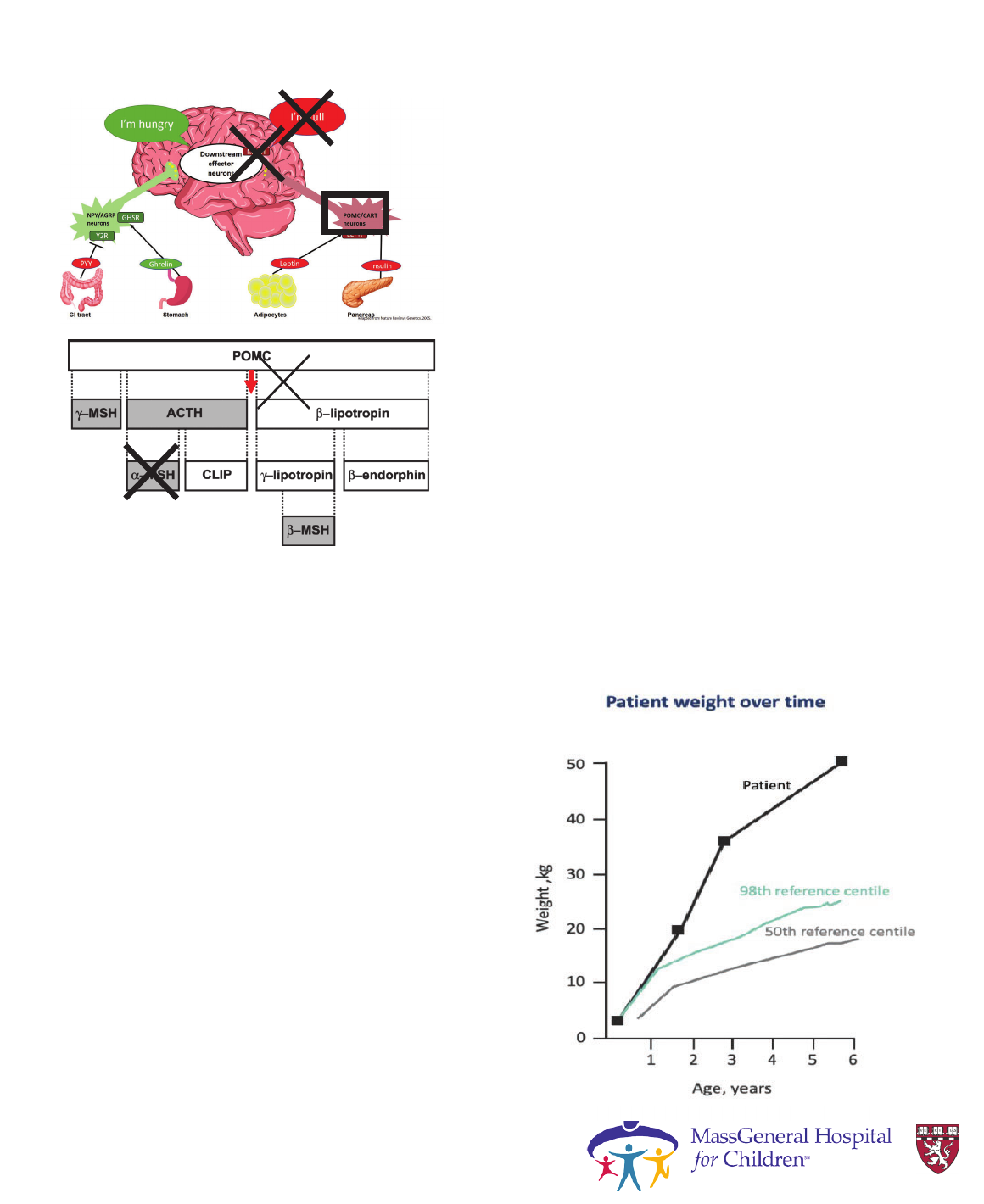
Gut-to-brain hunger signaling
AdipocytesGI tract Stomach Pancreas
I’m full
(anorexigenic)
Food intake
Energy Expenditure
NPY/AGRP
neurons
Ada
p
ted from Nature Reviews Genetics. 2005.
POMC/CART
neurons
LEPR
Y2R
GHSR
PYY
Leptin
Ghrelin
Insulin
Second order
neurons in PVN
hypothalamus
MC4R
Y1R
I’m hungry
(orexigenic)
Food intake
Energy Expenditure
LEPR
ɲ-MSH
POMC
BBS1-20
ALMS1
Courtesy Dr. Laurie
Braun
Objectives
• To discuss the energy balance regulation
• To introduce rare disorders of obesity, also known as monogenic
obesity.
• Review clinical features of syndromic genetic disorders that cause
obesity

Congenital Leptin
Deficiency
• Cause: mutations in gene for leptin (LEP)
• Phenotype: Hyperphagia with severe, early
onset obesity, altered immune function and
delayed puberty
• Prevalence: EXTREMELY RARE – only
several case reports in consanguineous
families*
• Diagnostic test: leptin level (undetectable),
genetic testing
• Treatment: recombinant leptin
*Mostly found in consanguineous families.
Adapted from Bell CG, et al. Nat Rev Genet. 2005;6(3):221-34.
Monogenic Obesity
Leptin deficiency
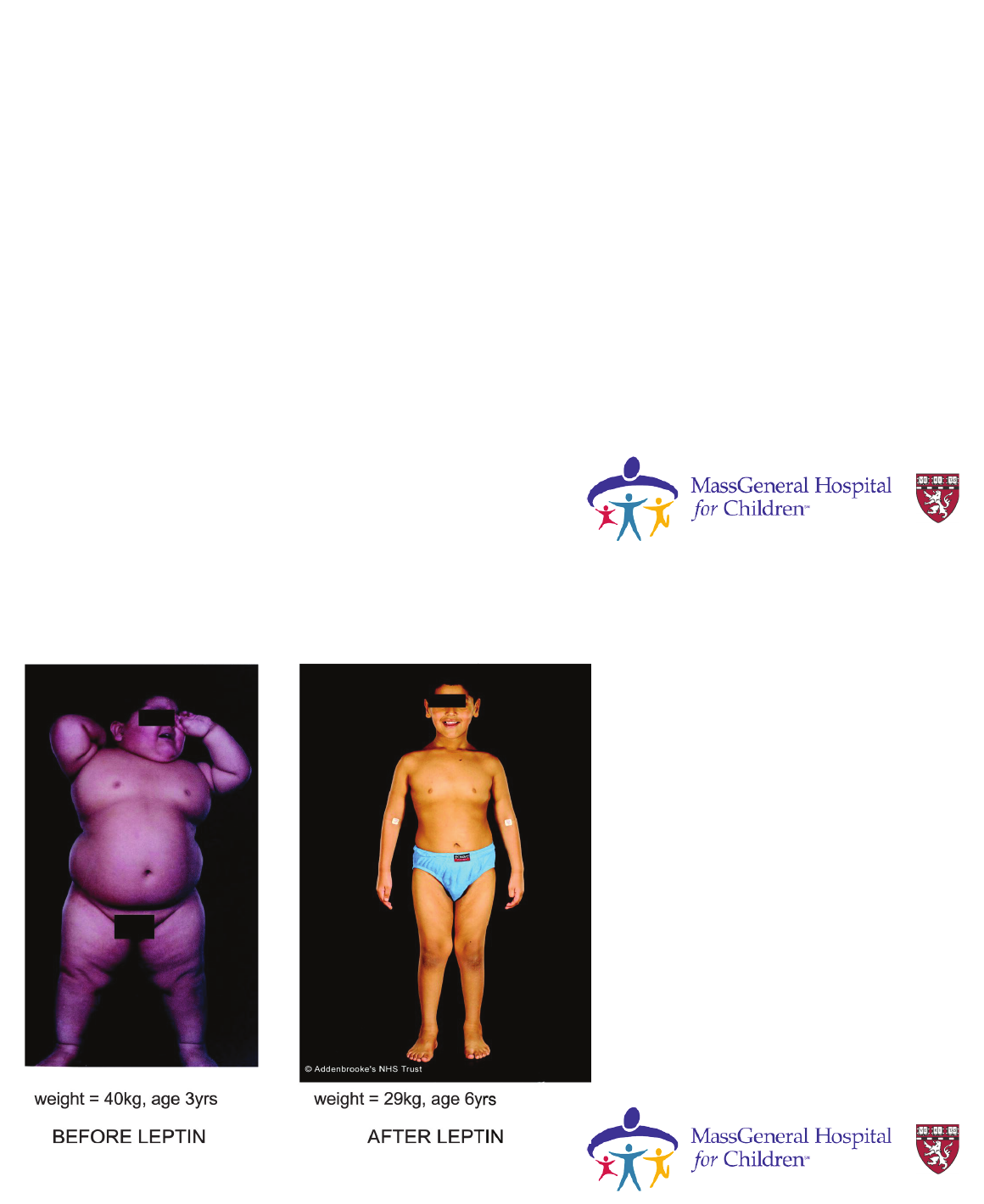
Case
• Female patient with rapid early onset
weight gain at 4 months of age
• Hyperphagia ( demanding food
continuously, ate much more than siblings)
• Developed growth abnormalities in leg
bones
-corrective leg surgery
-liposuction of lower limb fat to try to
improve mobility
Montague et al Nature. 1997;387:903–908
Leptin deficiency
Case continued..
• Endocrine tests
- Serum Leptin undetectable
- Insulin ( Elevated markedly)
- Proinsulin ( 4 times the ULN)
• Genetic tests:
- Homozygous LEP frameshift mutation detected
- Parents were heterozygous
• Eligible for treatment with recombinant human leptin replacement
Montague et al Nature. 1997;387:903–908
Leptin
treatment in
Leptin
deficiency
Source: Endocrinology Reviews
Leptin receptor
deficiency
• Cause: mutations in gene for leptin
receptor (LEPR)
• Phenotype: Hyperphagia with severe,
early-onset obesity and problems with
sexual development [same as leptin
deficiency]
• Prevalence: EXTREMELY RARE- only
several case reports*
• Diagnostic test: leptin level [very high],
genetic testing
• Treatment: none available, MC4R agonist
in development
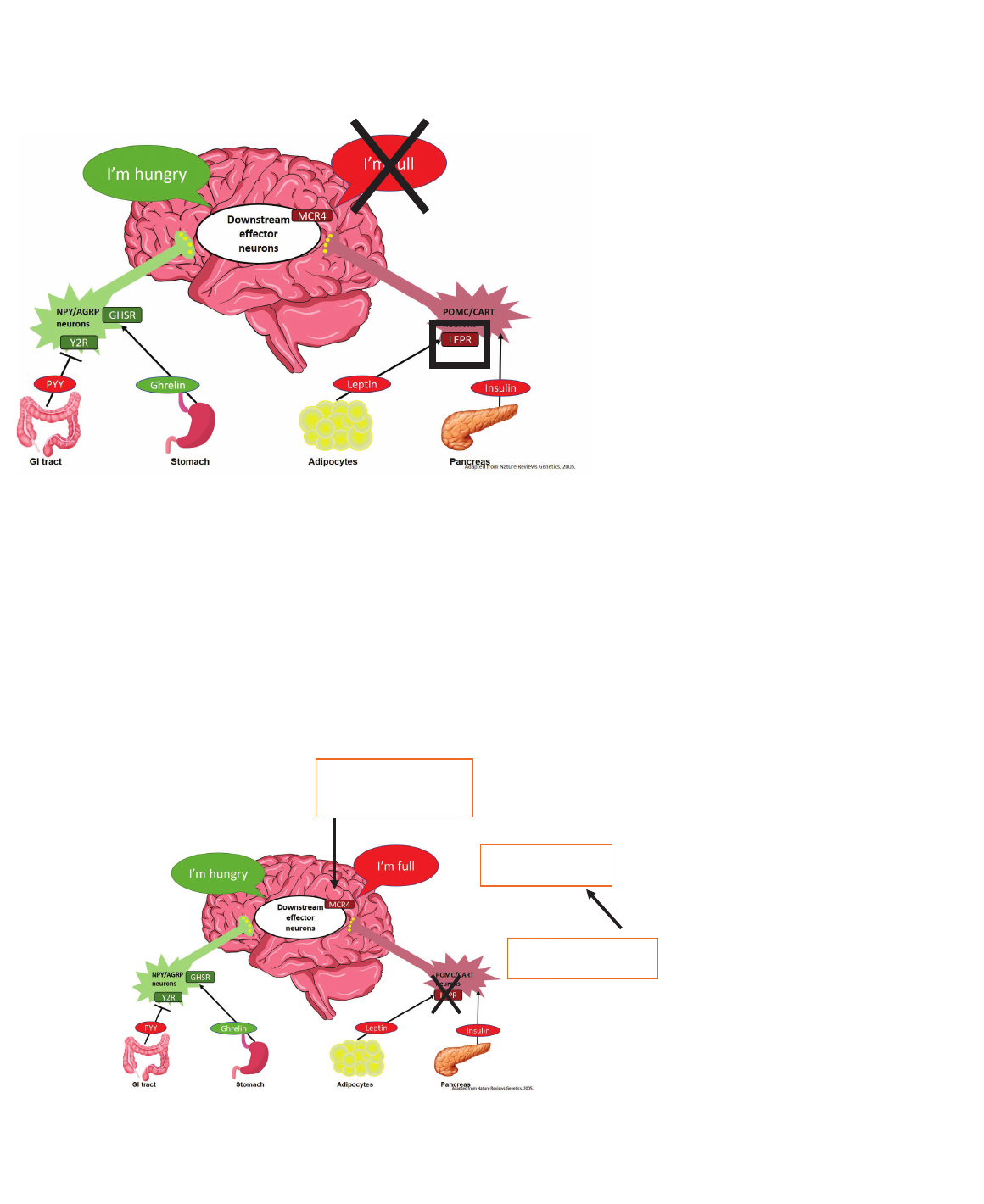
Monogenic Obesity:
Leptin Receptor Deficiency:
What’s Next?
Adapted from Bell CG, et al. Nat Rev Genet. 2005;6(3):221-34.
Impaired activation of
POMC neurons
Leading to lack of
MSH signaling
? Therefore, MC4R
agonist might be of
therapeutic benefit.

LEPR Deficiency
Case 2
• 2-year-old female presents with
progressive severe obesity from
birth
• Consoled only by food
• Parents were unable to maintain
nutritional plan owing to
hyperphagia
Kleinendorst L, et al. BMJ Case Rep. 2017;2017:bcr-2017-
221067.
Image reproduced from Kleinendorst L, et al. BMJ Case Rep.
2017;2017:bcr-2017-221067.
ih i i f blihi d
Case report: Leptin receptor
deficiency
• No developmental delays or other abnormal clinical features
present.
• Thyroid and cortisol levels normal
• Leptin levels elevated because of fat mass
• Sequenced MC4R and found no mutations
-Subsequently sequenced 52 obesity related genes.
• Found compound heterozygous LEPR mutations
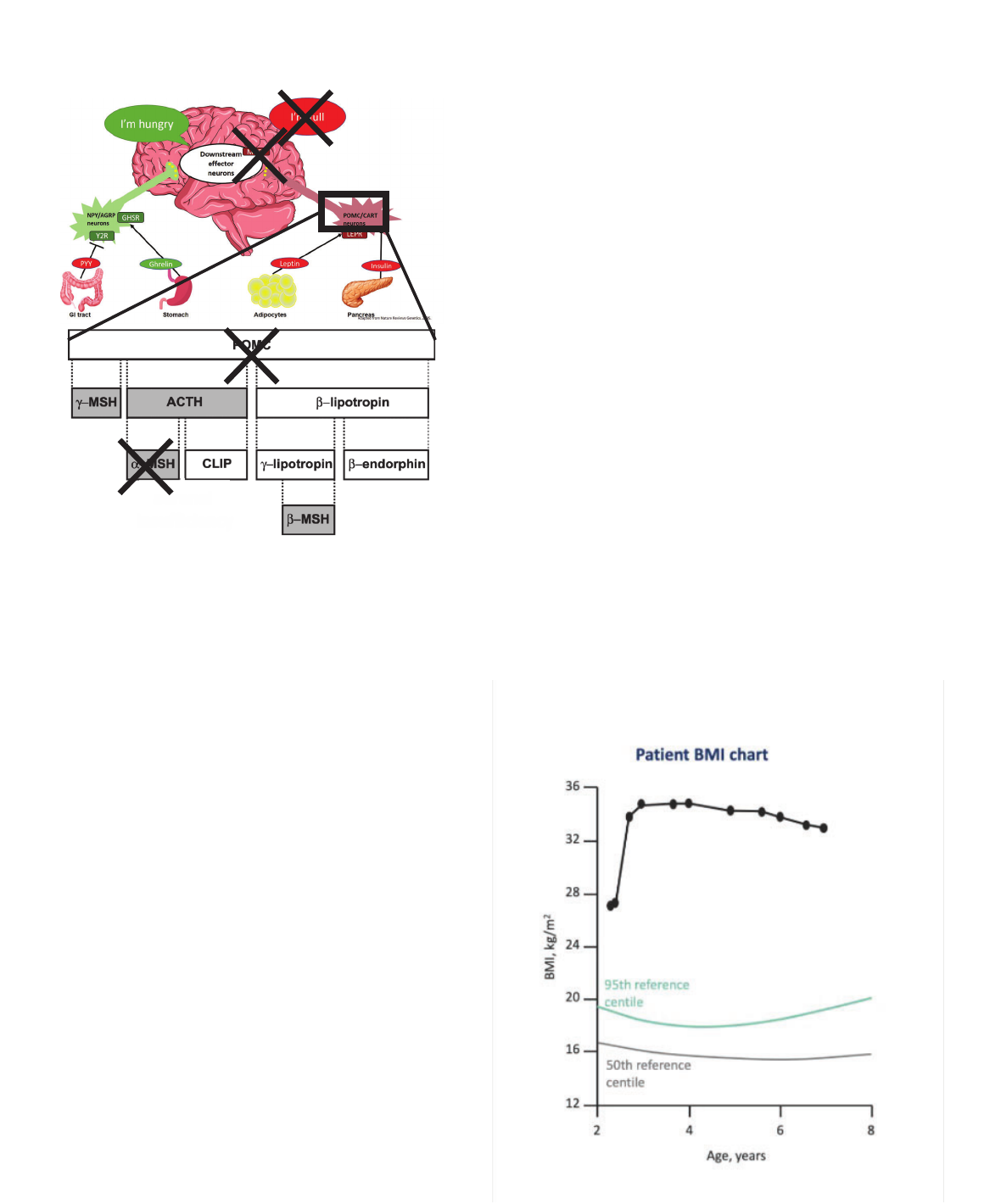
POMC mutations
(Pro-opiomelanocortin)
M
Monogenic Obesity:
• Cause: mutation in POMC gene
• Phenotype: early-onset obesity, adrenal
insufficiency, red hair
• Prevalence: <1/1,000,000
• Diagnostic test: distinct phenotype, low cortisol,
genetic testing
• Treatment:
o Hydrocortisone replacement for adrenal
insufficiency
Krude, Gruters, Trends in
Endocrinology & Metabolism, 2000.
adrenal
adrenal
insufficiency
red hair
POMC Deficiency:
Case Report
• 2-year-old Hispanic boy presents with
early onset severe obesity
-Neonatal Hypoglycemia
-Frequent respiratory infections
-Speech and motor delay
• Marked Hyperphagia
• Associated Adrenal Insufficiency and
hypothyroidism
-Hydrocortisone and levothyroxine
replacement
Hilado and Randhawa.J Pediatric Endocrinol Metab.2018;31:815-819
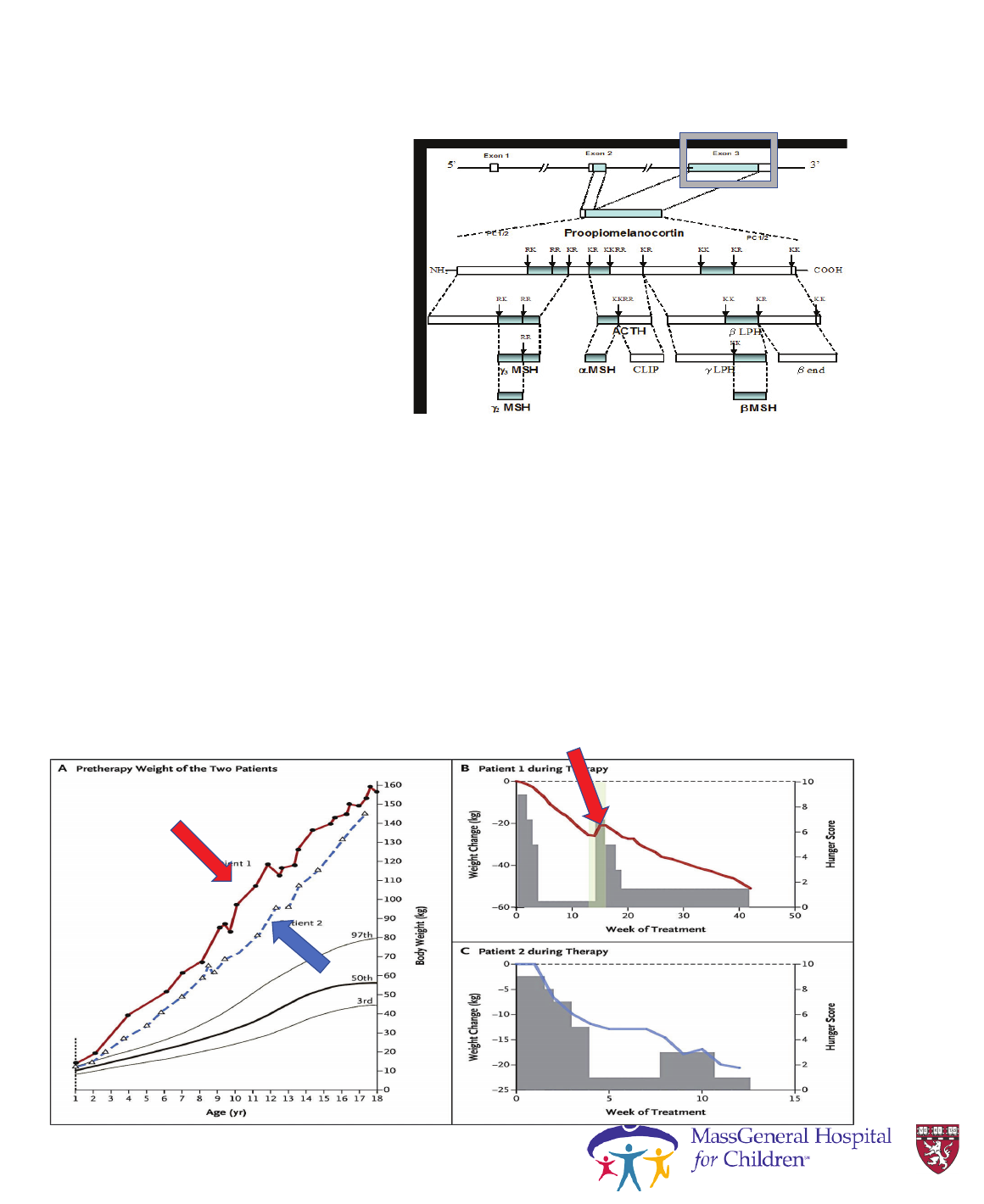
POMC Deficiency:
Case 3 (cont’d)
• Negative testing for Prader-
Willi syndrome
• Identified homozygous
POMC mutation in exon 3
• Patient was treated with
metformin
• Over a 3-year metformin
treatment span, BMI
decreased from 34.9
kg/m
2
to 32.9 kg/m
2
BMI = body mass index.
Millington GW. Nutr Metab (Lond). 2007;4:18. Open Access.
POMC Deficiency: What next? MC4R agonists
PC1/3 mutations
(Pro-hormone convertase)
M
Monogenic Obesity:
• Cause: mutation in PCSK1 gene
• Phenotype: severe obesity, low insulin, chronic
diarrhea, problems with sexual development
• Prevalence: VERY rare- only several case reports*
• Diagnostic test: high pro-hormone levels, genetic
testing
• Treatment:
o Hormone replacement
PC1/3
PCSK1 Deficiency
Case Report
• 6-year-old male with severe
early onset obesity
• Malabsorptive diarrhea noted
at the age of 8 days
• During first year of life required
specialized formulas for weight
gain
• Reported to be hyperphagic
with food seeking behavior at
the age of 2 .
Farooqi et al.JCEM 2007;92:3369-3373

PCSK1 Deficiency
Case 4 (cont’d)
• Proinsulin levels markedly elevated (1079 pmol/L Ref
range: <7 pmol/l), with insulin abnormally low.
• Low serum cortisol (as defects in cleaving POMC):
Treated with hydrocortisone
• Elevated ACTH precursor
(549 pmol/l Ref range:78 pmol/l)
• Low free T
4
T
4
= thyroxine.
Farooqi IS, et al. J Clin Endocrinol Metab. 2007;92(9):3369-73.
Clinical features of severe early onset obesity,
abnormal Insulin/proinsulin ratio and
sequencing diagnosed with prohormone
convertase (PC) 1/3 deficiency
MC4R mutationsM
Monogenic Obesity:
• Cause: mutations in MC4R receptor
(autosomal dominant)
• Phenotype: Normal mental status;
• Increased fat and lean mass with increased
bone mineral density
• Accelerated linear growth( tall stature)
• Hyperinsulinemia
• Prevalence: General population at 1:2000
• Prevalence in patients with obesity :0.5 to 1%
• Diagnosis : genetic testing
• Treatment: MC4R Agonist (Rhythm) in
development

Learning Objectives
• Review the energy balance regulation pathway.
• To introduce rare disorders of obesity, also known as monogenic
obesity.
• Review clinical features of syndromic genetic disorders that cause
obesity
S
Syndromic obesity
• The most frequent forms of syndromic obesity are Prader-Willi
and Bardet-Biedl syndrome.
• Not a single gene mutation but multiple genes are effected-- and have
more features besides just obesity
• Mechanism of obesity is less well understood
Prader-Willi Syndrome
Prader-Willi Syndrome
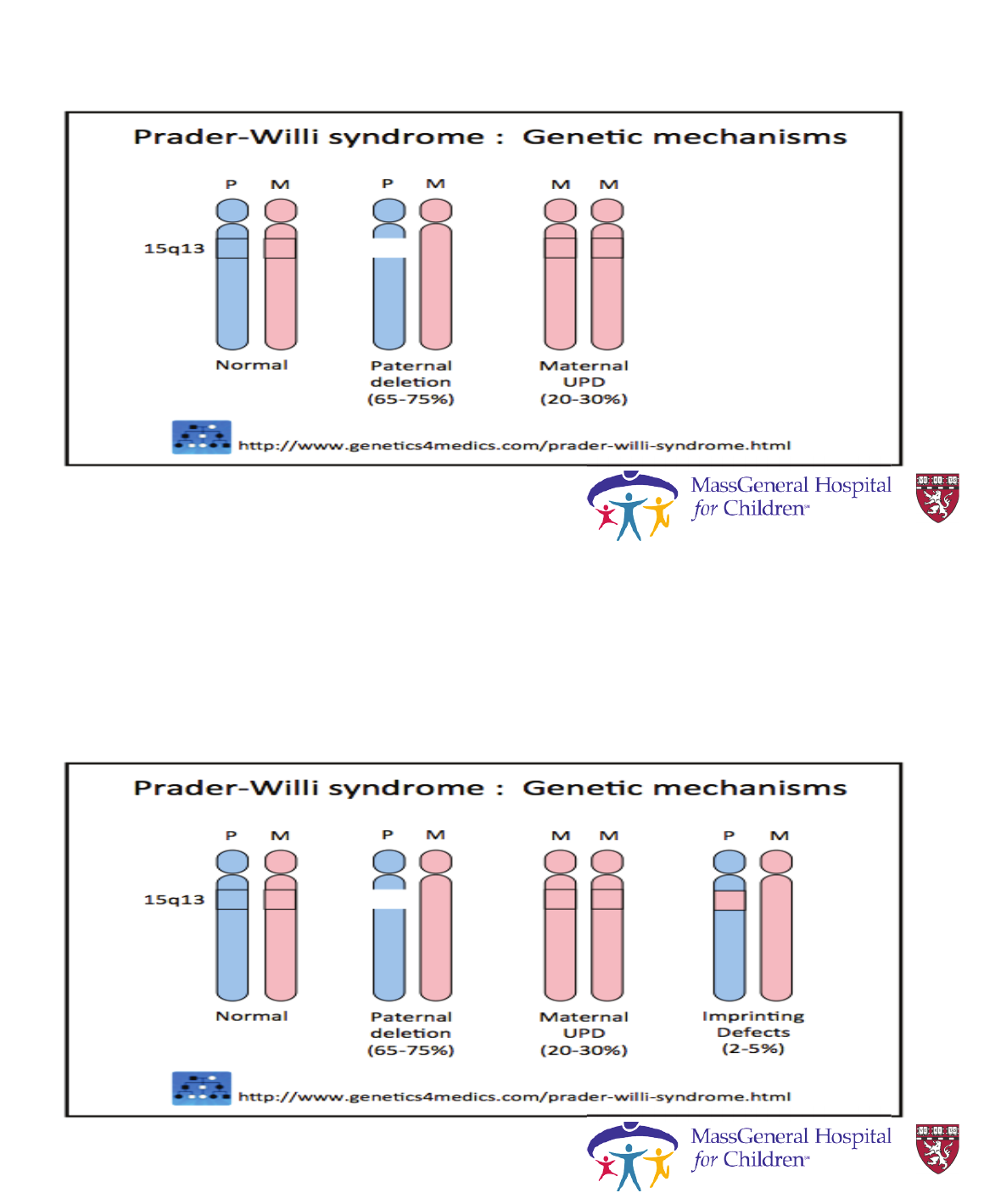
Prader-Willi Syndrome
Prader-Willi Syndrome
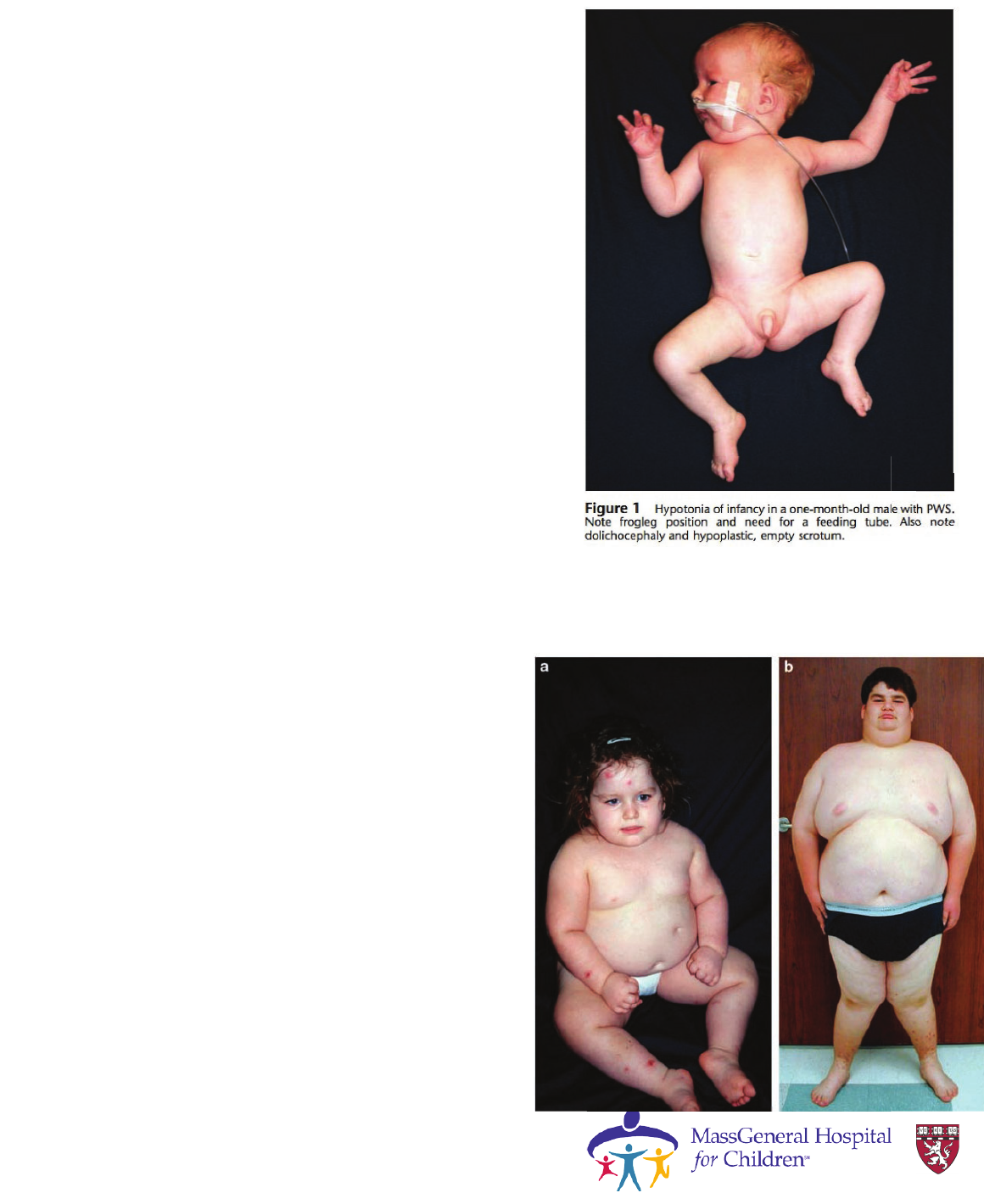
Clinical characteristics of
Prader-Willi Syndrome
• Birth to 2 years :
Hypotonia with poor suck
• 2–6 years
Hypotonia with poor suck
Global developmental delays
• 6–12 years :
History of hypotonia with poor suck
Global developmental delay
Excessive eating
(hyperphagia, obsession with food)
Central obesity
Cassidy, S., & Driscoll, D. (2009). EJHG, 17(1), 3-13.
Clinical characteristics of
Prader-Willi Syndrome
12 years through adulthood
Intellectual disability
Hyperphagia with central obesity
Hypothalamic hypogonadism
Typical behavior problems
(including temper tantrums and
compulsive features)
Cassidy, S., & Driscoll, D. (2009). EJHG, 17(1), 3-13.

Bardet-Biedl Syndrome (BBS)
• Mutation in BBS genes
• BBS genes are involved in
trafficking LEPR to the
neuronal cell surface
• Also genetic defect in cilia
Clinical Characteristics
• Diagnostic criteria : 4 primary features
OR 3 primary plus 2 secondary features
• Primary Criteria:
-Rod cone dystrophy
-Polydactyly
-Obesity
-Genital anomalies
- Renal anomalies
- Learning diabilities
Secondary Criteria:
- Speech delay
- Developmental
delay
- Diabetes mellitus
- Dental anomalies
-Congenital Heart
disease
-Brachydactyly
-Ataxia/Poor
coordination
- Anosmia/Hyposmia
Forsythe and Beales.Eur J Hum genet.2013;21:8-13

Alstrೌm syndrome: ALMS1 deficiency
• Mutation in ALMS1 gene.
• ALMS1 plays role in LEPR
signaling and POMC neuron
survival
• <1:1,000,000 (~900 cases)
Alstrೌm syndrome: Clinical Characteristics
v

Albright Hereditary Osteodystrophy
• Inactivating mutation in GNAS
Inherited from the mother, can
be associated with resistance to
certain hormones, in particular
the PTH. This is
Pseudohypoparathyroidism type
1A.
• When inherited from father , no
hormone resistance but an AHO
phenotype .
Albright Hereditary Osteodystrophy
phenotype
• Developmental delay
• Short stature
• Round facies
• Short fourth and fifth
metacarpals
• Brachydactyly
• Hypocalcemia ( in PHP1
A)

Fragile X Syndrome
• Obesity in upto 60% of
cases
• 1/2,500 births X-linked
• FMR1 gene (Xq27.3)
• Intellectual disability,
hyperkinetic behavior,
macroorchidism, large ears,
prominent jaw
Benefits of Identifying Genetic Cause
• Families relieved to know cause, feel less blame
• Anticipatory guidance and screening
• Social support groups provide community
• Management of hyperphagia as a physiologic medical condition
• Distinct approach from traditional nutritional counseling for obesity
• Food dosed and timed similar to prescription medications
• In hypotonic conditions, 20% to 40% lower caloric needs due to decreased lean mass
• Growth hormone therapy approved for PWS – increases muscle mass and tone, reduces truncal
obesity, potential cognitive benefits when initiated early
• Genetic diagnosis o opportunity for targeted treatment
Styne DM, et al. J Clin Endocrinol Metab. 2017;102(3):709-57; Rubin DA, et al. Food Nutr Res. 2015;59:29427;
Goldstone AP, et al. .J Clin Endocrinol Metab. 2008;93(11):4183-97

• Leptin replacement for leptin deficiency
• Melanocortin agonist (setmelanotide) under investigation for
LEPR, POMC, PCSK1, and CPE mutations
• Targeted approaches being studied for other rare and common
variants of leptin pathway genes
Precision Medicine Based on Genetics
Farooqi IS, et al. J Endocrinol. 2014;223(1):T63-70.
Kühnen P, et al. N Engl J Med. 2016;375(3):240-6.
Clement K, et al. Nat Med. 2018;24(5):551-5.
